
(9 pm. – promoted by ek hornbeck)
NMR (Nuclear Magnetic Resonance) spectrometry is one of the big guns in organic chemistry and biochemistry for determining how atoms are strung together in molecules. There are many different kinds, but the two of greatest utility to chemists are proton NMR and carbon-13 NMR. A friend of mine asked me for some help for his daughter who is studying the subject in Organic Chemistry right now, so I thought that I might as well use it as a topic for this series.
Before we get deep into the subject, note that some authors refer to NMR spectrometry and others to NMR spectroscopy. I prefer the former term because the connotation of spectroscopy, to me at least, has to do with lenses, prisms, and diffraction gratings, making it an optical method. There are no analogous devices in NMR, so I prefer spectrometry.
All NMR has some features in common, so we might as well cover the basics first. By the way, this has nothing to do with nuclear energy, and the only radiation present is in the radiofrequency range, so it will not fry you.
This is heavily connected with quantum mechanics, but I shall try to use analogies that are more easily visualized than a bunch of equations. I do not intend for this to be a graduate level abstract.
It turns out that some atomic nuclei behave as if they are spinning magnets. Moving magnets create an electric current, and electric currents are affected by external magnetic fields. In NMR, we take advantage of these facts and expose the nuclei (in whole molecules) to a strong magnetic field, then bombard them with radiofrequency energy. It turns out, because of quantum mechanics, these nuclei can have only specific energies, and when one absorbs a radiofrequency photon, it will be promoted from a lower energy level to a higher one. Since we can measure these absorptions very precisely, we can tell when they have been promoted.
The nuclei of most use are ones that a spin quantum number of +/-1/2. This is because, in a given magnetic field, there are only two states possible, one where the magnetic moment is aligned WITH the external magnetic field, and the other where it is aligned AGAINST the field. We call these parallel and antiparallel configurations. The parallel one is just a tiny bit less energetic than the antiparallel one. It turns out that the two elements most important in organic chemistry are carbon and hydrogen, and both of those have isotopes whose nuclei half integer in spin.
There are a couple of other concepts that are important. One is the magnetogyric ratio, which is merely how strongly a nucleus interacts with the external magnetic field. The larger the magnetogyrc ratio, the stronger the interaction and thus, everything else being equal, the more sensitive the nucleus to analysis. Another factor that contributes to sensitivity is the relative abundance of the nucleus compared with other nuclei that have a spin of zero. In hydrogen, way over 99% of the nuclei have half integer spin. In addition, these protons have a magnetogyric ratio of 267.513 γ / 106 rad s−1 T−1. The units are not important for this discussion. Carbon-13 has a magnetogyric ratio of 67.262 γ / 106 rad s−1 T−1, or only 25.1% of protons. That means if the same number of nuclei were present, the carbon-13 would give only a quarter of the signal of the protons.
Here is a nice overview of NMR with some graphics that illustrate some of the concepts that I am explaining in this piece:
But it gets worse. Carbon-13 occurs at only 1.1% natural abundance, the rest being the nonactive carbon-12. So, at natural abundance, carbon-13 is only 2.77 x 10-3 as protons. Before the advent of high speed computing devices, it was just not feasible to use carbon-13 NMR. All of this has changed.
So why is NMR so valuable? It has to do with two things. If all protons (or carbons) in a molecule were in exactly the same environment, it would have no value. However, it turns out that many more times than not they are NOT all the same, and these tiny differences affect the energy difference betwixt the parallel and the antiparallel states. We are talking TINY differences, but easily measured ones. Simplistically, it has to do with how much electron density is around a given proton or carbon-13. This is called shielding, and nuclei that are more shielded resonate at a lower energy than those that are less shielded, because the electron density “hides”, or shields, them from the external magnetic field, making the external field have less effect on them, thus making the energy gap larger. This means that unless there are elements of symmetry in the molecule, most of the nuclei are in different electronic environments.
There is another interaction that is critical as well. This is called spin-spin coupling, and it affects what the signals look like. This is critically important to determine how many hydrogens are bonded to a given carbon, and this information along with the energy needed to cause a transition gives not only what kinds of protons and carbon-13 nuclei, but how they are connected. Let us look at an example.
This is a nice picture of the structural formula of ethanol, beverage alcohol, and an actual proton NMR spectrum of it.
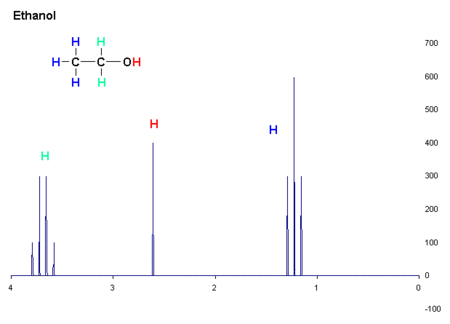
In proton NMR, hydrogens connected to carbons adjacent to other carbons bonded to hydrogens interact with each other and perturb the energies in predictable ways. Note that the signal from the three hydrogens on the -CH3 group are split into a triplet by the two hydrogens on the adjacent -CH2 group. Those are in turn split into a quartet by the three hydrogens on the methyl group. This is quite general. The hydrogen bonded to the oxygen does not participate because of rapid exchange with other hydrogens bonded to oxygen on nearby molecules. These hydrogens move from molecule to molecule too fast to interact with the other five on the NMR timeframe, which is fairly slow. In proton NMR the distance of the splitting of the peaks is a very useful guide to molecular geometry and are greatly used. There are lots of tables of these coupling constants, and along with the chemical shift are key data.
In general, coupling constants are not as important in carbon NMR, for a couple of reasons. One is that they tend to be very large compared to those in proton NMR, and become confounding as we will see later. Another is that in general the coupling in carbon NMR is only betwixt the hydrogens bonded directly to a specific carbon, so little geometric information is encoded. Indeed, techniques have been developed to reduce or eliminate coupling for carbon NMR.
Now, if you have fooled around with NMR very much you immediately recognize this as ethanol. The rules as we know them say that -CH2 hydrogens resonate at a higher energy (lower field) than -CH3 ones. Hydrogen bonded to oxygen can be all over the place, but its position in the picture is typical. Do you see the numbers below the x axis of the graph? Those are called delta units, and they have no physical dimensions. They are used because everything is referenced from zero, on the right. They are measurements of the energy difference betwixt a given proton and one that, by convention, resonates at zero. The position to the left (almost always) of zero is called the chemical shift, and it is strongly dependent on the electronic environment of the nuclei being probed. Over many years of study, there are tables and tables of chemical shifts.
Now that we have had a primer into proton NMR, let us turn to carbon NMR. But first, we need to review some traditional terms that stick with us but do not reflect current practice like they used to do. In the old days, for several technical reasons, it was easier to use a narrowband, pure, almost single frequency of RF energy and use coils to modify the magnetic field. Those were the old permanent magnet instruments like the Varian EM-360, one of which I was able to modify to perform some pretty exciting stuff. Ask me about CIDNP in the comments! Anyway, nuclei with higher deltas resonate at a lower field (higher energy), and anything to the left of zero was called a downfield shift. These continuous wave (CW) instruments are still useful for lots of routine analyses, because they are rugged, reliable, and cheap. A typical scan for proton NMR took about five minutes or so, but since carbon-13 is only 2.77 x 10-3, a similar scan would take 1805 minutes, or over 30 hours. Actually, for some technical reasons it most often would have taken much longer than that.
More modern instruments use superconducting magnets, so now it is easier to play around with the RF frequency than to perturb the magnetic field, but the term still is used. What is done now is that the external field is held constant, with a lock compound added to the sample acting as a control. Then, every second or every few seconds usually, a broadband RF pulse is passed into the sample. The instrument then “listens” for the decay of the signal, and that decay is the combination of all of the nuclei interacting with the pulse. The pulse is wide enough in bandwidth to excite all of the nuclei at once, and the information is encoded in the free induction decay (FID) signal. I looks like a squiggly line, and to the human eye is completely unintelligible. That is because the FID is a time domain signal, and a typical NMR spectrum is a frequency domain signal. Thus, the signal has to be “transformed” from time space to frequency space. This was not possible for self contained instruments until comparatively recently, to do this a Fourier transform is required. It was only in the 1970s that this was possible in self contained instruments, and most of them were research and development devices. In the 1980s, computer technology improved, and it was possible to perform the FTs on the fly, leading to the terminology Fast Fourier Transform (FFT) instruments. Before that, the individual FIDs had to be recorded on tape and processed later.
The trend towards superconducting magnets is because the stronger the magnetic field, the greater the energy difference betwixt the parallel and antiparallel states. The greater the energy difference, the greater the resolution of the instrument. There is another reason for higher fields, and that is when the energy difference betwixt the two states is greater, it is harder to saturate the higher level. Saturation occurs when the population of nuclei in the upper level becomes equal to that of the lower level, and then no more RF energy can be absorbed, and the signal vanishes. Actually, what really happens is that as many hihg energy state nuclei emit the same rf energy as lower state absorb it, so the net signal is zero. It turns out that it takes time for the energy to be radiated so that the nucleus can return to the lower energy state. This is called the relaxation time, and in general carbon-13 has longer relaxation times than protons. This is the reason why a CW scan for carbon-13 NMR would actually take much longer than the 30 hours mentioned a while ago.
All carbon-13 NMR is now done by FFT pulse instruments. It is just not practical otherwise, but who knows where future technology will lead? In modern practice, a sample is placed in a very uniform tube and placed betwixt the pole faces of the superconducting magnet. By the way, car keys, jewelery of any kind, cell phones, and credit cards are not allowed in a high field NMR room because of the extremely high fields involved. I have heard horror stories about metal chairs flying about the room when the superconducting, high field magnet was first charged, and I believe the source to be reliable. The tube is spun at a fairly high speed, because even the best magnets are not perfect, and since NMR has a fairly slow timeframe, this evens out the inhomogeneities in the field.
Once a lock is made, the actual analysis begins. Pulse after pulse of RF energy is passed through the sample, the FID acquired, and the FFT done in the blink of an eye. You can actually sit and watch the spectrum grow into something recognizable. Depending on the concentration of the analyte, a decent carbon-13 trace can be acquired in just a few minutes, not the 30 plus hours if using CW techniques.
But it gets even better. Remember spin-spin coupling from a while ago? This does not appear betwixt carbons, because the probability of two adjacent ones, since carbon-13 is only at 1.1% natural abundance, so the likelihood of two carbon-13 nuclei being adjacent is only 0.0001%. But it DOES occur betwixt the hydrogens that are connected to a given carbon, and can be a mixed blessing. It turns out the the coupling constants are very large, and sort of interfere with making sense of the spectrum. But we have a remedy. There will be more on that in a bit.
First, we need to get an idea about the chemical shifts of different kinds of carbon. Here is a nice chart that illustrates them.
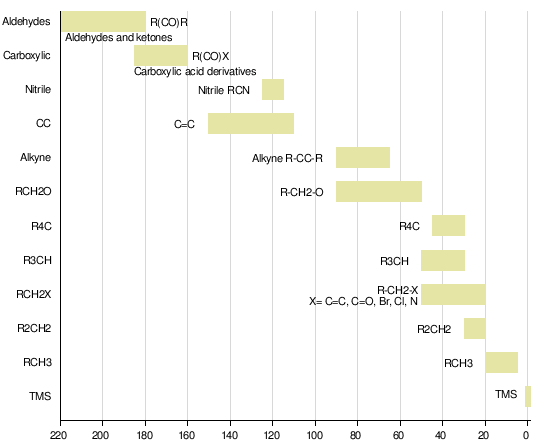
The one at delta = 0 is for TMS, tetramethylsilane, almost universally used as the reference material except in systems in which it is not soluble. You can see that there is a good spread in chemical shifts, so C-13 NMR is quite valuable in that many signal peaks are well separated.
There are three basic way to collect carbon NMR signals. One is allow the protons that might be on each carbon to have a full spin/spin interaction (this method is called fully coupled). However, except for the simplest molecules these spectra can be extremely complicated and thus difficult to interpret.In the next method, called off resonance decoupling, a weak rf signal corresponding to nearly the resonance frequency of protons in superimposed on the rf pulse that causes the carbons to resonate, and removes much, but not all, of the coupling from protons on the carbons. This allows information to be gained about how many protons are connected to each carbon without having a really cluttered spectrum.
The third way to collect the signals is to use broad band decoupling, where a strong rf signal that includes all of the proton frequencies is superimposed on the carbon resonance frequency. This causes all of the multiplets to collapse to singlets, greatly simplifying the spectra. Some graphic examples are in order, both it illustrate these concepts and to show you how to interpret carbon-13 nmr spectra.
First, let us look at the fully coupled spectrum of 1,2,2-trichloropropane. Here is a hand drawing of the structural formula, because I could not find one online that explicitly showed the hydrogens:
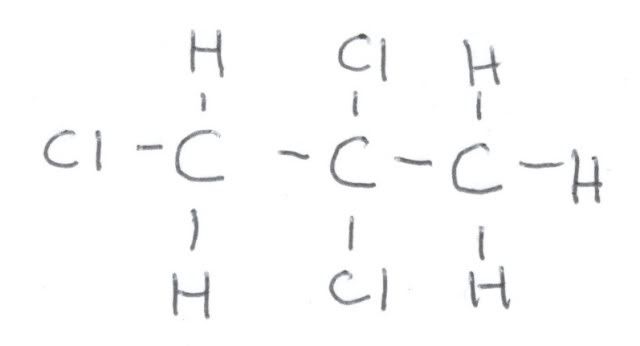
Here is fully coupled spectrum of the material. At zero ppm is the reference, TMS, and it is a quartet because each carbon is equivalent and is bonded to three hydrogens. It is NOT part of the 1,2,2-trichloropropane. At about 32 ppm we see a quartet, and that means that this carbon is attached to three hydrogens. Looking at the formula, that must be the one on the very right. If we look at the correlation table, we would expect a methyl group to be closer to 20 ppm, but the chlorines adjacent to it deshield it somewhat, causing it to resonate at lower field. At around 60 ppm we see a triplet, and that means that this carbon is bonded to 2 hydrogens. That means that this is the carbon on the very left. This is still a little lower field that the correlation table indicates, but the chlorines adjacent and bonded to that carbon cause that. Finally, at around 85 ppm is a singlet, which must arise from the center carbon since it is not bonded to a hydrogen.
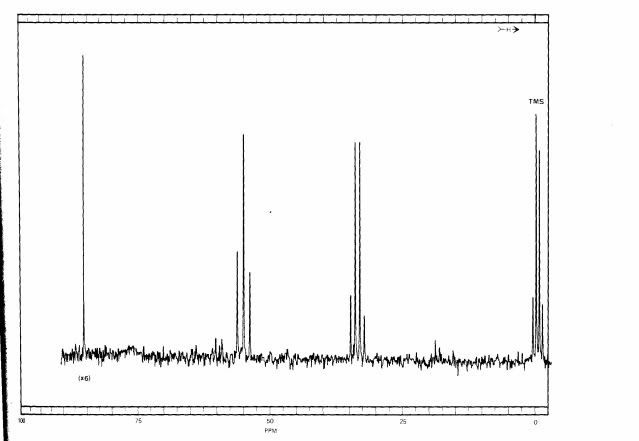
Here is the fully decoupled spectrum:
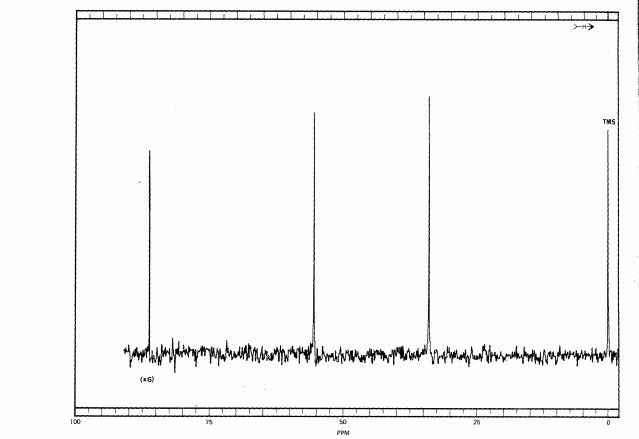
As you can see, the information about how many hydrogens are attached to the carbons is lost. In simple systems like this, the loss of information can be a disadvantage, but coming up we can see why fully decoupled spectra are often preferred.
Here are the fully decoupled and the fully coupled spectra of a related compound, and we shall decide what it is merely from the spectrum:
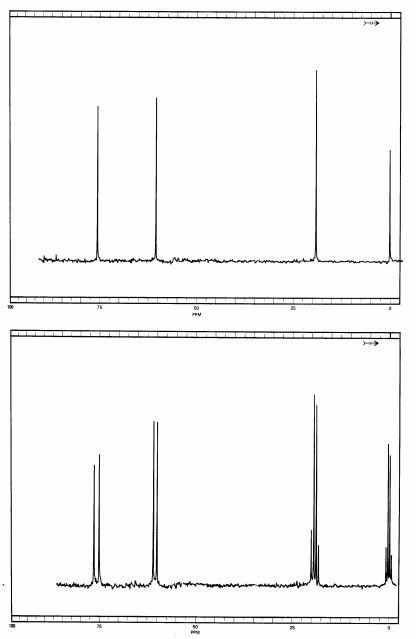
In the fully decoupled spectrum, you will see that it looks a whole lot like the 1,2,2 compound, and without more information, such as running authentic samples of each, it would be hard to decide which spectrum belongs to what. But look at the fully coupled spectrum.
Aside from TMS, we see a quartet at 20 ppm, and that must be a carbon with three hydrogens on in. Next, at 60 ppm we have a doublet, meaning that this carbon must be bonded to a single hydrogen. At around 77 ppm there is another doublet, so there must be a second carbon bonded to a single hydrogen. We can also make a couple of other observations. The methyl group in this compound is at a higher field than on the 1,2,2 one, and that can be explained by speculating that the methyl group is adjacent to fewer chlorine atoms (which decrease electron density on neighboring atoms, deshielding them). The doublet at around 60 ppm is around the same chemical shift as the triplet on the 1,2,2 compound, so me might speculate that this carbon has only one chlorine on it. Finally, the triplet at around 75 ppm is fairly close in chemical shift to the singlet for the 1,2,2 material.
So that means we have a methyl group bonded to a carbon that carries only one chlorine. That carbon must be bonded to another carbon carrying two chlorines. Since a methyl group is by definition only at a terminus of a molecule, there is only possibility that is in keeping with all of the data. This compound is 1,1,2-trichloropropane, with the following structure:
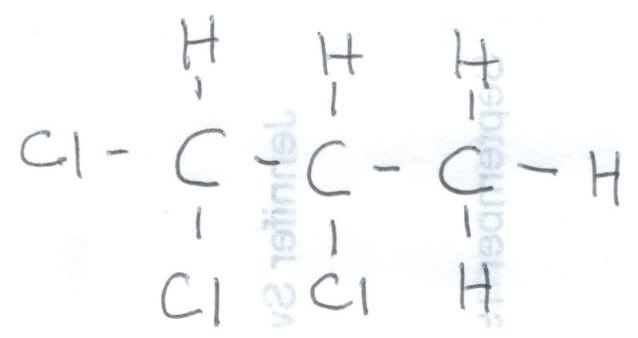
Now for a more complex system, that of ethyl phenylacetate. The fully coupled spectrum is very complex, and if that were all that you had to go on, it would be hard to interpret it, because we can not even count carbons like in the simpler cases earlier:
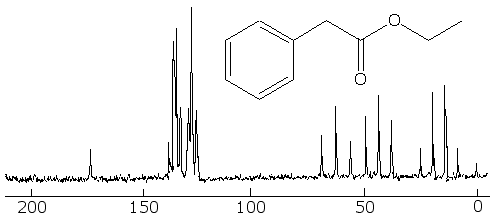
But in the fully decoupled one we can count the different kinds of carbon in it, and that makes things easier. First, we see that there are 8 different kinds of carbon (remember not to count TMS at zero). Looking at the correlation table, we identify the one at around 10 to 15 ppm as a methyl group. The one around 40 ppm is likely a CH2 group attached to the methyl group, and the one at 60 ppm is the CH2 bonded betwixt the aromatic ring and the carbonyl group. The four peaks betwixt about 130 and 140 ppm are the four kinds of aromatic carbons on the ring, but it is not easy to say which ones belong to each peak. The one at 175 ppm is certainly the carbonyl carbon.
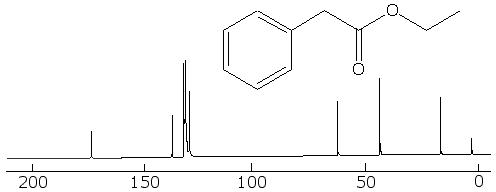
With this information, we can look at the fully coupled spectrum again and make sense out of it. Sure enough, the 15 ppm peak is a quartet, so it must be a methyl group. The ones at 40 and 60 ppm are triplets, so they are indeed methylene groups. The aromatic carbons are still not distinguishable, but the carbon at 175 ppm has to be the carbonyl one, because it is a singlet.
Here is the off resonance decoupled spectrum of the same material:
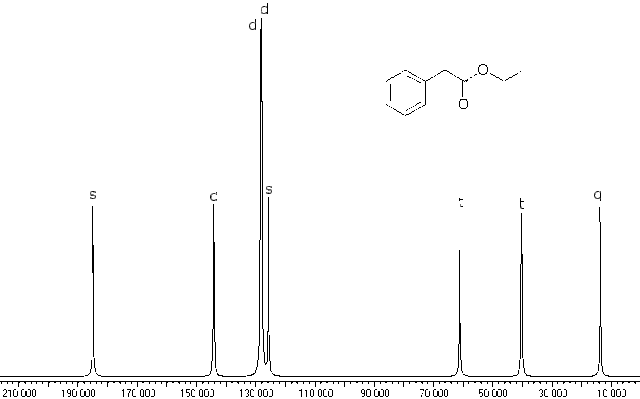
At first glance it looks a lot like the fully decoupled one, but the computer adds letter codes for the multiplicity of the peaks and even though it is not visible to the eye, the computer can record the information. You can see the letters “s”, “d” (sort of garbled), “d”, “d”, “t”, “t”, and “q” reading from left to right. We still can not determine which doublets at 130 to 150 ppm belong to which aromatic carbons, but this does confirm that we correctly assigned them as aromatic ones earlier. It DOES indicate that the aromatic resonance at 124 ppm is the carbon on the aromatic ring bonded to the side chain, for otherwise it would be a doublet. Since it is not bonded to hydrogen, it is a singlet.
This has been a very brief introduction to carbon-13 NMR, and there is a tremendous amount of additional information that is way beyond the scope of this article. My point here was to present something that nontechnical people could understand and even have a little fun with, sort of like solving puzzles. Another goal was to illustrate the importance of NMR in chemical analysis. The final goal was to present something with enough detail to help my friend’s daughter make some sense out of it for her Organic Chemistry course in college. Hopefully, I have succeeded in all three areas. I am sure that there will be lots of comments and questions.
There are many other analytical techniques in the chemist’s arsenal, and they give different information about molecular structure. Among them are infrared absorption spectroscopy, mass spectrometry, UV-Vis spectroscopy, electron spin resonance spectrometry, and a host of others, including the so called “hyphenated” techniques like gas chromatography/mass spectrometry.
Well, you have done it again! You have wasted many more einsteins of perfectly good photons reading this magnetic piece. And even though Newt Gingrich again realizes that the will never become President of the United States when he reads me say it, I always learn much more writing this series than I could ever possibly hope to teach. Thus, please keep those comments, questions, corrections, and other feedback coming. Tips and recs are also very welcome. I shall stay around as long as comments warrant (if something good does not happen) and shall return tomorrow evening for Review Time to clean up any comments that I might miss tonight.
Warmest regards,
Doc, aka Dr. David W. Smith
Crossposted at
Daily Kos, and
2 comments
Author
solving puzzles?
Warmest regards,
Doc
Author
I very much appreciate it.
Warmest regards,
Doc